V. Các tổn thương liên quan đến cơ
1. Cơ nâng đòn (levator claviculae muscle)
Cơ nâng đòn là một cơ bình thường ở 2% đến 3% dân chúng. Cơ mọc từ các củ trước của các mỏm ngang đốt sống cổ trên, chạy xuống giữa cổ tới cơ ức đòn chũm và bám vào 1/3 giữa hoặc 1/2 ngoài xương đòn (Hình 26). Nó có thể ở một bên hoặc hai bên. Điều quan trọng về hình ảnh của cơ này là nhận ra nó là một cấu trúc bình thường chứ không phải là hạch bạch huyết to, mạch máu không ngấm thuốc, hoặc các khối khác [89,90].

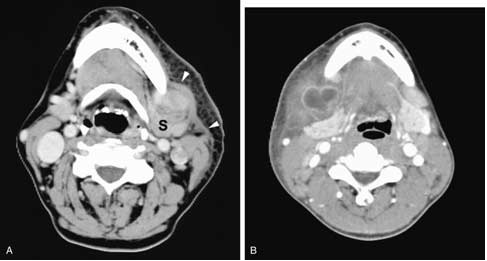
Hình 26. CT + C cổ trên (A) dưới (B) cho thấy cơ nâng đòn bên trái. Cơ nằm sau cơ ức đòn chũm ở giữa cổ và bên ngoài nó ở cổ thấp.
(CT + C = CT tiêm thuốc cản quang)
2. Teo cơ sau cắt dây thần kinh (Denervation Atrophy)
Cắt dây thần kinh cơ dẫn tới các thay đổi cuối cung như cơ bị teo, nhiều vùng cơ bị mỡ thay thế. Giai đoạn mới cắt có phù thần kinh khiến cơ to ra và cường độ tín hiệu cao trên ảnh T2W [91,92]. Giai đoạn mạn tính đánh giá tốt bằng T1W, biểu hiện là bó cơ teo với các vùng lan tỏa tăng tín hiệu tương ứng với thay thế mỡ. Hình ảnh trên CT cũng tương tự nhưng không rõ bằng MRI. Tốc độ phát triển rất biến đổi, các dấu hiệu phù tồn tại cho tới 48 tháng sau cắt dây thần kinh hoặc thay đổi mỡ ngay từ tháng thứ hai [92]. Cả CT và MRI chỉ thấy mỡ ở giai đoạn cuối mà không có các sợi cơ hình 27.
 Hình 27. Một loạt các ảnh CT từ trên (A) xuống dưới (D) ở bệnh nhân phẫu thuật nền sọ với tổn thương các dây V, VII, IX và XII. (A) teo rõ cơ ức đòn chũm phải (đầu mũi tên) và thay thế mỡ của các cơ lưỡi bên phải. Hậu quả, đáy phải của lưỡi bị sa so với bên trái, các cơ cắn và cơ chân bướm trong cũng nhỏ hơn các cơ cùng tên bên trái. (B) teo cơ ức đòn chũm phải (đầu mũi tên) các cơ thang. Cũng có teo các cơ ở bên phải sàn miệng. (C) teo rõ cơ ức đòn chũm phải (đầu mũi tên) và cơ thang (mũi tên). (D) CT qua vùng cổ thấp ở một bệnh nhân khác đã được phẫu tích rễ cổ trái cho thấy teo cơ thang trái (đầu mũi tên). Cơ thang phải bình thường, nhưng bó cơ có thể nhầm với khối (mũi tên).
Hình 27. Một loạt các ảnh CT từ trên (A) xuống dưới (D) ở bệnh nhân phẫu thuật nền sọ với tổn thương các dây V, VII, IX và XII. (A) teo rõ cơ ức đòn chũm phải (đầu mũi tên) và thay thế mỡ của các cơ lưỡi bên phải. Hậu quả, đáy phải của lưỡi bị sa so với bên trái, các cơ cắn và cơ chân bướm trong cũng nhỏ hơn các cơ cùng tên bên trái. (B) teo cơ ức đòn chũm phải (đầu mũi tên) các cơ thang. Cũng có teo các cơ ở bên phải sàn miệng. (C) teo rõ cơ ức đòn chũm phải (đầu mũi tên) và cơ thang (mũi tên). (D) CT qua vùng cổ thấp ở một bệnh nhân khác đã được phẫu tích rễ cổ trái cho thấy teo cơ thang trái (đầu mũi tên). Cơ thang phải bình thường, nhưng bó cơ có thể nhầm với khối (mũi tên).
3. Phì đại cơ
Phì đại cơ có thể phát triển như là kết quả của luyện tập, kết hợp với các rối loạn tăng trương lực (tăng hoạt động tự phát), hoặc một cách tự phát (một số trường hợp phì đại cơ cắn). Trong phì đại cơ thực sự, cơ to ra nhưng hình ảnh bình thường. Cũng có giả phì đại cơ do thâm nhiễm cơ bởi các bệnh loạn dưỡng cơ, amyloidosis, sarcoidosis, bệnh tuyến giáp và biểu hiện của ký sinh. Sau cùng, viêm cơ có thể làm cơ to ra. Trên CT, cơ phì đại có thể xuất hiện bình thường về tỷ trọng, nhưng cường độ tín hiệu MR bất thường thấy ở hầu hết các cơ to do giả phì đại hoặc viêm cơ.
Một thí dụ khá phổ biến của phì đại cơ luyện tập ở đầu và cổ xảy ra với phì đại cơ cắn và ở các bệnh nhân sau phẫu tích rễ cổ hình 28. Sau phẫu thuật, vai sa xuống sau khi cắt thần kinh của cơ thang; do đó phần lớn các bệnh nhân phục hồi cơ nâng vai cùng bên, cơ này có thể giả khối và thường bị chẩn đoán nhầm là một khối u tái phát. Tương tự, các người nâng vật nặng thường phát triển phì đại cơ vùng cổ thấp, lưng trên và các cơ vai, xu hướng cân đối.

Hình 28. T1W (A) cho thấy cơ cắn hai bên to ra (M) ở bệnh nhân có phì đại cơ cắn. (B) CT + C ở một bệnh nhân đã cắt rễ cổ cho thấy teo cơ thang trái (đầu mũi tên). Cơ nâng vai trái phì đại (mũi tên).
4. Fibromatosis colli (sternomastoid or sternocleidomastoid tumor)
Vẹo cổ (torticollis) có thể do bẩm sinh hoặc mắc phải. Nguyên nhân có thể là cơ, thần kinh, nhiễm khuẩn, u, sau chấn thương, hoặc thậm chí tâm lý. Khi vẹo cổ do thâm nhiễm xơ keo cơ ức đòn chũm bởi một khối có thể sờ thấy ở cổ trẻ mới sinh, bệnh được gọi là fibromatosis colli (khối u xơ ở cổ) hoặc sternocleidomastoid tumor (khối u cơ ức – đòn – chũm). Bệnh này phải phân biệt với vẹo cổ do cơ (muscular torticollis) là một bệnh kéo căng cơ mà không có khối. Tuy nhiên, 15% tới 20% các bệnh nhân có khối u xơ ở cổ (fibromatosis colli) tiến triển dần thành vẹo cổ do cơ cho dù được điều trị [61]. Hiếm khi fibromatosis colli biểu hiện ở hai bên. 70% các trường hợp diễn ra từ lúc sinh đến tháng tuổi thứ hai, chủ yếu ở bên phải. Phần lớn các khối u loại này thoái triển tự phát mà không có hậu quả biến dạng, và phương pháp điều trị bảo tồn được lựa chọn. Có nhiều giải thích, nhưng nguyên nhân chính xác không rõ [93,94].
Khối u cơ ức-đòn-chũm (sternocleidomastoid tumor) biểu hiện là sưng phồng tổ chức xơ, rắn, lành tính liên quan chủ yếu ở 1/3 giữa hoặc 1/3 dưới của cơ ức đòn chũm hình 29. Khối giả u này xảy ra ở 0,4% trẻ sơ sinh, hầu hết thoái triển hoàn toàn sau vài tháng tiếp theo. Trên siêu âm, khối u xơ thường xuất hiện dưới dạng một khối tăng âm hoặc cơ ức đòn chũm to ra lan tỏa với đậm độ âm hỗn hợp; các hình thái khác hiếm xảy ra. Mô mềm xung quanh vẫn bình thường [95]. Mặc dù đặc điểm tín hiệu so với cơ bình thường có thể biến đổi, cường độ tín hiệu trong fibromatosis hơi tăng trên T2W và tăng rõ trên gradient-recalled T1W; cường độ tín hiệu trên SE-T1W bình thường [96].

Hình 29. CT (A) cho thấy cơ ức đòn chũm trái to ra, tỷ trọng hơi thấp so với cơ bình thường. T1W + C (B) ở một bệnh nhân khác cho thấy cơ ức đòn chũm to, nổi rõ (mũi tên). Cả hai bệnh nhân này có fibromatosis colli.
5. Vẹo cổ do cơ (muscular torticollis)
Về bệnh học, vẹo cổ do cơ (muscular torticollis) biểu hiện là sự tăng sinh nguyên bào xơ và/hoặc keo có giới hạn không tạo ra khối. Như vậy nó có thể được xem là thuộc phổ của fibromatosis colli, mặc dù 73% các muscular torticollis xuất hiện sau 1 tuổi. Nếu nó biểu hiện trước 6 tháng tuổi, sự kéo căng thụ động có thể có ích. Sau tuổi đó, có thể phải cắt gân [61].
Vẹo cổ bẩm sinh do cơ, hay vẹo cổ co cứng tự phát (idiopathic spasmodic torticollis) nguyên nhân có thể không biết hoặc liên quan đến chấn thương hoặc thấy trong nhiều bệnh lý cột sống, nội sọ, bệnh lý đốt trục – đốt đội (atlantoaxial), cột sống [94,97-125]. Do sự kết hợp của vẹo cổ bẩm sinh do cơ với tư thế bất thường của đầu trong tử cung, nó được qui là gây chấn thương cơ ức – đòn – chũm, dẫn đến hội chứng giống kiểu hội chứng khoang (compartment – type syndrome) [117]. Singer và cộng sự nghiên cứu các bệnh nhân vẹo cổ bằng CT và MRI đã phát hiện các dấu hiệu gợi ý của xơ hóa trong cơ ức đòn chũm được chứng thực khi phẫu thuật [113]. Mặc dù congenital mascular torticollis thường được phát hiện ở trẻ còn ẵm ngửa và trẻ nhỏ, nó cũng xuất hiện ở thanh niên và người trưởng thành trẻ. Nên chẩn đoán phân biệt với các nguyên nhân tư thế bất thường của đầu không rối loạn trương lực. Trong nghiên cứu một loạt các bệnh nhi có vẹo cổ cấp, gồm một số không có tiền sử chấn thương, 2/3 (13/21 bệnh nhân) có trật xoay nhẹ đốt trục-đốt đội (AARS) trên CT [126]. Người ta gợi ý rằng vẹo cổ liên quan đến trật xoay nhẹ đốt trục-đốt đội có thể không được giải quyết đầy đủ cho đến khi bán trật này được chú tâm điều trị, hoặc sửa chữa dưới hướng dẫn của chiếu X quang hoặc bằng nối (fusion) [127].
Năm 1969, Snyder đã mô tả vẹo cổ bộc phát (paroxysmal torticollis) ở trẻ 2 đến 8 tháng tuổi. Các đợt vẹo đầu kéo dài từ vài giờ tới 3 ngày và đôi khi kèm theo xanh xao, nôn, hoảng sợ lúc bắt đầu cơn. Với hầu hết các trẻ này các cơn chấm dứt tự phát vào tuổi lên 5 [128]. Bệnh này có thể mang tính gia đình, và 12% bệnh nhân với mất thăng bằng bộc phát thời kỳ thơ ấu diễn ra trước đó đã có vẹo cổ bộc phát ở thời kỳ ẵm ngửa [129].
Đánh giá vẹo cổ thường được thực hiện bằng siêu âm cho thấy cơ ức đòn chũm to ra lan toả. Bất thường có thể giảm âm hoặc đồng âm, phụ thuộc vào tuổi của tổn thương. Mặc dù cơ to ra có thể nhầm với một khối, nó luôn chuyển động cùng với cơ trên siêu âm thời gian thực [130]. Bởi vì luôn có dấu hiệu siêu âm bất thường ở bệnh nhân vẹo cổ do u xơ (fibromatosis torticolli), do đó nên thực hiệu siêu âm ở các bệnh nhân có nghi ngờ bệnh này [131].
Trên CT, cơ ức đòn chũm to ra là đồng tỷ trọng với cơ bình thường, hoặc đồng nhất hoặc hơi không đồng nhất. Trên MRI, cơ có thể đồng tín hiệu hoặc tín hiệu thấp hơn cơ bình thường, hoặc đồng nhất hoặc hơi không đồng nhất, và phần chân của cơ bị ảnh hưởng nhiều hơn phần đầu. Điều quan trọng là loại trừ các khối ở cổ như nang khe mang, nang bạch huyết và các bệnh ác tính như sacôm cơ vân (rhabdomyosarcoma) và u nguyên bào thần kinh (neuroblastoma) [130].
6. Viêm cơ (myositis)
Cơ vân có thể viêm sau một viêm mô tế bào và/hoặc áp xe lân cận. Chính trong trường hợp này thuật ngữ myositis được dùng. Cơ sưng và nổi rõ (ngâm thuốc) sau tiêm thuốc đối quang trên cả CT và MRI hình 30,31. Trên MRI, cơ có cường độ tín hiệu T2W cao hơn cơ bình thường, và có viêm các cấu trúc lân cận.
Thuật ngữ viêm cơ tăng sinh (proliferative myositis) mô tả một tổn thương giả sacôm của cơ vân phát triển nhanh. Ở đầu và cổ, nó thường xảy ra ở cơ ức đòn chũm. Nó có xu hướng diễn ra ở các bệnh nhân nhiều tuổi, không gặp ở trẻ em. Hiếm hơn, các ổ dạng xương hoặc xương xuất hiện, có thể chữa khỏi bằng tiểu phẫu [61].
Viêm cơ khu trú (focal myositis) là giả u do viêm của cơ vân. Nó phát triển nhanh và xảy ra ở một cơ. Cả người lớn và trẻ em đều bị. Hiếm khi xảy ra ở đầu và cổ nhưng đã được báo cáo đối với cơ ức đòn chũm, vùng dưới cằm, vùng miệng. Chữa khỏi bằng phẫu thuật.

Hình 30. CT + C cho thấy viêm mô tế bào ở cổ trái. Có dầy da, mỡ dưới da tăng tỷ trọng và rộng, dầy cơ bám da cổ (mũi tên) là kết quả của myositis.

Hình 31. T1W (A) và T2W (B) cho thấy dầy nhẹ phần trước cơ ức đòn chũm trái và một vùng cường độ tín hiệu T2W cao. Bệnh nhân có viêm cấp tuyến nước bọt dưới hàm trái và viêm mô tế bào. Bệnh nhân đã được điều trị kháng sinh, và mặc dù các dấu hiệu khác của nhiễm khuẩn đã bớt đi, anh ta vẫn đau cơ ức đòn chũm. Đây là viêm cơ (inflammatory myositis).
7. Viêm cơ cốt hóa (myositis ossificans)
Viêm cơ cốt hóa (myositis ossificans) có hai dạng. Dạng phổ biến là myositis ossificans circumscripta (MOC, viêm cơ cốt hóa khu trú), thường là tiến triển khu trú xảy ra ở ngoài vùng đầu và cổ. Dạng hiếm hơn và lan tỏa hơn là myositis ossificans progressiva (viêm cơ cốt hóa tiến triển). Viêm cơ cốt hóa khu trú (MOC) là một bệnh không hay gặp, xảy ra sau chấn thương, bỏng, nhiễm khuẩn và ở các bệnh nhân với bệnh lý không biết. Mặc dù nguyên nhân vẫn còn chưa được biết, phần lớn các nhà điều tra tin rằng MOC biểu hiện một quá trình phản ứng khu trú không sinh u gây tăng sinh mô trung mô không biệt hóa, mô này thâm nhiễm vào mô mềm bao quanh. Các nguyên nhân được gợi ý bao gồm cốt hóa bọc máu tụ, rách màng xương khiến các tế bào dưới màng xương xâm lấn vào cơ, dị sản cơ và mô liên kết. Mặc dù MOC ít xảy ra ở đầu và cổ; nó đã được báo cáo ở cơ cắn, cơ chân bướm trong, cơ thái dương, cơ mút, cơ cằm móng, cơ ức đòn chũm và các cơ cạnh cột sống. MOC thường xảy ra ở người trẻ, chơi thể thao. Trong các trường hợp xác định được tiền sử chấn thương, thời gian từ khi chấn thương tới khi phát hiện lâm sàng là 3 tuần tới 20 năm. Các bằng chứng X quang của vôi hóa trong MOC có thể thấy từ 2 đến 4 tuần và hóa xương ngay từ tháng thứ 4 đến tháng thứ 5 [132].
Trên CT, viêm cơ cốt hóa khu trú (MOC) có vỏ vôi hóa ngoại vi và/hoặc cốt hóa ở trong một khối khu trú. Tuy nhiên, vôi hóa rất sớm có thể nhầm là ngấm thuốc cản quang, đặc biệt nếu protocol chụp CT không có cả thì trước và sau tiêm thuốc. Các hình ảnh giai đoạn sớm cũng gợi ý một khối u và viêm lân cận. Hình thái này phù hợp với mô tả mô học sớm của tăng sinh nguyên bào xơ trung tâm và nguyên bào xơ cơ với phù phối hợp hình 32 [133, 134], chúng có thể cản trở sự phân biệt mô học của MOC với bệnh ác tính. Do đó, khả năng của MOC nên thông báo cho nhà giải phẫu bệnh nếu thực hiện sinh thiết.
Ba giai đoạn tiến triển của MOC đã được mô tả: giai đoạn giả viêm, giai đoạn giả u, và giai đoạn mạn tính. Dấu hiệu hình ảnh phụ thuộc vào giai đoạn của bệnh. Trên T2W, các tổn thương sớm biểu hiện điển hình là một khối mô mềm không đồng nhất với phù bao quanh lan rộng. Các ảnh tiêm đối quang từ thường thấy khối nổi rõ. Các tổn thương đã chín được xác định rõ hơn, không có phù. Tuy nhiên hình ảnh MRI không có khả năng loại trừ một bệnh ác tính.
Hình ảnh MR của các tổn thương đã chín cho thấy khối không đồng nhất, ranh giới rõ với cường độ tín hiệu gần bằng cường độ tín hiệu của mỡ trên cả T1W và T2W, viền giảm tín hiệu và không có phù bao quanh hình 33 Trên MRI, chẩn đoán phân biệt gồm áp xe sớm, hạch hoại tử và u. Hình ảnh của pyomyositis và polymyositis cũng có thể giả tổn thương MOC giai đoạn sớm trên cả CT và MRI. Nếu hút kim nhỏ để chẩn đoán MOC, cần lấy thêm một mẫu ở ngoại vi [132].

Hình 32. CT + C (A và B) bệnh nhân 38 tuổi 1 tuần sau đau cổ. (A), một vùng lờ mờ tỷ trọng thấp ở trong các cơ cạnh cột sống trái to ra (mũi tên trắng), có một đường cong mờ (mũi tên đen) có thể diễn giải là ngấm thuốc trong khối. Cắt về phía dưới (B) cho thấy các cơ cạnh sống trái to và tỷ trọng thấp không rõ ràng (mũi tên). CT không tiêm thuốc cản quang 1 tháng sau đợt đau (C và D) cho thấy ở (C) có đám cốt hóa/vôi hóa (mũi tên đen) trong các cơ cạnh sống trái to ra, tỷ trọng thấp (mũi tên trắng). Vùng tỷ trọng cao này tương ứng với đường cong tỷ trọng cao trong hình A. Trong hình D, các cơ cạnh sống trái to ra, tỷ trọng thấp.

Hình 32 tiếp. CT (E và F) 4 tháng sau đợt đau cho thấy ở E đám vôi hóa/cốt hóa vẫn còn; tuy nhiên tỷ trọng thấp trong các cơ cạnh sống trái không còn. (F) tiêm thuốc cản quang cho thấy các cơ cạnh sống cân đối. Các dấu hiệu này minh hoạ tiến triển của myositis osssificans.

Hình 33. Cùng một bệnh nhân ở hình 31. MRI chụp 1 tuần sau đau cổ. (A) T1W, có một khối mô mềm ranh giới không rõ (mũi tên), trong khi ở (B) T2W khối (mũi tên) có hình trông tín hiệu được hiểu là mạch máu hoặc xơ trong tổn thương. Vùng trung gian có tín hiệu cao kiểu phù. (C) ảnh T2W ở phía dưới hơn, cơ và mô mềm kề cận to ra và cường độ tín hiệu cao (mũi tên). (D) T2W một tháng sau khởi bệnh, khối và phù trong các cơ cạnh sống đã giảm. Bệnh nhân này có viêm cơ cốt hóa (myositis ossificans).
8. Loạn dưỡng cơ (Muscular Dystrophies)
Các loạn dưỡng cơ (muscular dystrophies) là một nhóm các bệnh nguyên phát của cơ có bản chất di truyền. Các khiếm khuyết hóa sinh cơ bản không được mô tả rõ. Trong các loạn dưỡng, một bệnh chủ yếu tác động đến cơ cổ là loạn dưỡng mạc vai-cánh tay (fascioscapulohumeral dystrophy, FSHD). Các loạn ngoại lệ (tách khỏi nhóm), chúng nằm cùng một phổ nằm giữa các loạn dưỡng cơ của Ducheme và Becker, tác động tới các cơ gấp của cổ. Bệnh nặng đến mức bệnh nhân không thể nhấc đầu lên được, yếu chân xảy ra sau 12 tuổi. Các loạn dưỡng khác, như loạn dưỡng cơ của Emery-Dreifuss và loạn dưỡng cơ vai-mác (scapuloperoneal dystrophy), tác động đến các cơ vai và có thể thay đổi các cơ cổ.
FSH dystrophy là một bệnh di truyền trội nhiễm sắc thể thường. Tỷ lệ bệnh 1/100.000. Yếu cơ mặt thường nhận ra lần đầu vào thập niên thứ 2 của cuộc đời. Yếu vai đặc biệt tác động đến các cơ cố định bả vai. Yếu chân cũng có thể xảy ra. Dạng nặng hơn của bệnh xảy ra ở trẻ còn ẵm ngửa. Điều trị hỗ trợ [135].
Trên CT, các cơ bị thay thế mỡ hoàn toàn hình 34,35. Trên MRI, cơ có cường độ tín hiệu giống mỡ, và cường độ tín hiệu cơ bình thường không có.

Hình 34. CT + C trên (A) va dưới (B) ở vùng cổ cho thấy các cơ teo chứa đầy mỡ ở lưng trên và sàn của tam giác cổ sau. Bệnh nhân có fascioscapulohumeral dystrophy.

Hình 35. Chụp serie CT cổ từ trên (A) xuống (C) cho thấy sự thay thế mỡ của các cơ lưng, sàn của tam giác cổ sau, và vai phải. Bệnh nhân có fascioscapulohumeral dystrophy.
9. Loãn dưỡng tăng trương lực cơ (Myotonic Dystrophy)
Loạn dưỡng tăng trương lực cơ (là một trong các loạn dưỡng phổ biến nhất, tỷ lệ mắc 2,5 tới 5/100.000 dân. Cứ 1/20.000 đến 1/7.500 người mang sự đột biến gen đối với bệnh này. Hội chứng di truyền trội từ mẹ với mức độ biến đổi. Bất thường này đã được lập bản đồ là chuỗi lặp ba mẫu tự (trinucleotide) CTG trên nhánh q13.3 của nhiễm sắc thể 19, và khi bệnh di truyền từ mẹ thì mức độ thường nặng. Sự di truyền từ cha là hiếm [136,137].
Điển hình bệnh xuất hiện ở thập niên thứ hai tới thứ tư của cuộc đời; tuy nhiên trẻ em cũng có thể bị mắc. Thông thường, các cơ nhỏ của bàn tay và các cơ duỗi của cánh tay bị teo trước tiên. Tuy nhiên ở một số bệnh nhân, teo cơ mặt và sa mi mắt là các dấu hiệu khởi đầu. Thường cơ cắn và cơ ức đòn chũm bị tác động, dẫn tới biến dạng hàm và “cổ thiên nga”. Sự kết hợp của hói trán và các nếp nhăn trán làm cho bệnh nhân có một vẻ đặc biệt. Gần như tất cả các cơ đều có thể bị tác động, và phần lớn bệnh nhân phải ngồi xe lăn hoặc nằm giường ở vào lứa tuổi 15 đến 20. Tăng trương lực cơ có thể xảy ra trước yếu cơ hình 36. Mờ thủy tinh thể là phổ biến.
Bệnh cũng phối hợp với chứng dày xương ảnh hưởng đến xương sọ và nền sọ. Các dấu hiệu CT gồm có dày xương ở bản xương sọ, phần chũm xương thái dương, tháp xương đá, các mỏm yên trước, lưng yên và dốc nền (clivus). Bệnh liên quan đến ổ chảo của khớp thái dương hàm có thể gây trật khớp. Các thay đổi xơ hóa, tiêu và không phồng xương cũng có thể thấy ở xương thái dương [138,139]. Các bệnh nhân này có thể có xoang trán khổng lồ và hố yên nhỏ. Các dấu hiệu ít phổ biến khác gồm có vẹo đầu, góc mũi nhỏ, hẹp các ống tai trong, hai mắt gần nhau (hypotelorism), xương hàm to. Các thay đổi đó có thể ở một hoặc cả hai bên. Nguyên nhân của những dấu hiệu về xương này vẫn chưa biết [140].

Hình 36. CT cổ từ trên (A) xuống (C) cho thấy thoái hóa chứa đầy mỡ của các cơ ức đòn chũm và cơ thang, và mức độ ít hơn ở các cơ nền của tam giác sau. Cũng có giãn thực quản cổ và bệnh nhân có ống nội khí quản. Bệnh nhân này bị myotonic dystrophy.
10. Sacôm cơ vân (Rhabdomyosarcoma)
Rhabdomyosarcoma (RMS), u ác tính của cơ vân, chiếm 8% tới 19% tất cả sacôm mô mềm, và 35% tới 45% sacôm ở vùng đầu và cổ. Hầu như tất cả các khối u (98% tới 99%) chứa cơ vân là sacôm cơ vân, với phần nhỏ còn lại là u cơ vân (rhabdomyoma) [74]. Sacôm cơ vân (RMS) chủ yếu là một khối u của trẻ con và người trưởng thành trẻ, với hầu hết các trường hợp xảy ra ở bệnh nhân dưới 12 tuổi và 43% ở trẻ dưới 5 tuổi. Nó là sacôm phổ biến nhất ở trẻ em và đứng thứ bảy trong các bệnh ác tính của trẻ em sau leukemia, các khối u hệ thần kinh trung ương, lymphoma, u nguyên bào thần kinh (neuroblastoma), Wilms’ tumor, và ung thư xương [61]. Các thể mô học bao gồm thể phôi (embryonal), nang (alveolar), đa hình (pleomorphic), chùm (botryoid) và thể hỗn hợp. Hầu hết các nhà giải phẫu bệnh coi sacôm cơ vân hỗn hợp (mixed RMS) gồm hai hoặc nhiều thể mô học nói trên [75].
Hơn một nửa các sacôm cơ vân là thể phôi (embryonal) và 70% đến 90% loại này mọc ở đầu cổ hoặc đường niệu. Phần lớn gặp ở trẻ dưới 10 tuổi; tuy nhiên chỉ 1/4 các trường hợp diễn ra trong thập niên thứ hai. Di căn hạch vùng xảy ra ở 10% tới 38% các trường hợp [61].
Sacôm cơ vân hình nang (alveolar RMS) là loại phổ biến thứ hai, chiếm 1/4 các trường hợp RMS. Nó xảy ra chủ yếu ở bệnh nhân 15 đến 25 tuổi và tác động chủ yếu đến các chi và thân mình. Chỉ 18% các trường hợp xảy ra ở đầu và cổ. Loại này hay di căn hạch vùng (33%) và di căn hạch xa (75% đến 85%) [61, 75].
Sacôm cơ vân hình chùm (botryoid RMS) chỉ khác sacôm cơ vân thể phôi (embryonal RMS) ở hình thái đại thể. Khoảng 75% các khối u này mọc ở âm đạo, tiền liệt tuyến, hoặc bàng quang. Các khối u còn lại mọc ở đầu và cổ, hoặc đường mật. Hầu hết bệnh nhân từ 2 đến 5 tuổi.
Sacôm cơ vân đa hình (pleomorphic RMS) chủ yếu thấy ở bệnh nhân 40-60 tuổi, chỉ 6% được phát hiện ở trẻ dưới 15 tuổi. Hầu hết các trường hợp mọc ở chi, chỉ 7% mọc ở đầu và cổ. Di căn mạch máu là chủ yếu, chỉ có 9% bệnh nhân có di căn hạch vùng [61]. Thời gian sống sót kém nhất trong các loại sacôm cơ vân [75].
Về lâm sàng, sacôm cơ vân thường là một khối cứng, to nhanh. Do xạ trị và hóa liệu pháp, tiên lượng của bệnh nhân đã được cải thiện nhiều (hiện nay là 85% hoặc hơn) [74]. Tiên lượng của u liên quan chặt chẽ tới sự lan rộng của u hơn loại mô học. Theo vị trí, u ở ổ mắt có tiên lượng tốt hơn các vị trí ở cổ (thời gian sống sót 5 năm là 85% so với 55%) [74] do có khuynh hướng khu trú và ít di căn hạch.
Về hình ảnh, sacôm cơ vân giống một khối u thâm nhiễm dày đặc tế bào hình 37. Cơ vân luôn thấy tại ví trí gốc. Trên cả CT và MRI, các khối u điển hình là đồng nhất, phá hủy xương kề cận. Hoại tử có thể thấy, nhưng chảy máu trong u hiếm thấy, và điển hình là không có vôi hóa. Khối u đồng tín hiệu với cơ trên T1W và tăng tín hiệu trên T2W [142]. Hơn một nửa các trường hợp ngấm thuốc đối quang (nổi rõ) đồng nhất, và mức độ nổi rõ tương tự với cơ vân bình thường. Trong phần lớn các trường hợp thì bờ kém rõ.

Hình 37. T1W + C cho thấy khối có bờ kém rõ, nổi rõ sau tiêm, mọc ở các cơ cạnh sống bên trái cổ. Có xâm lấn mặt trái cột sống. Bệnh nhân có RMS.
11. Di căn tới cơ
Di căn theo đường máu tới cơ là hiếm. Tỷ lệ mắc tương đối thấp có thể liên quan đến các chất ức chế emzyme của cơ, môi trường acid của cơ và tác động ép mạch máu của co cơ [143]. Thường di căn từ khối u nguyên phát của phổi, đại tràng, tụy, vú, hoặc thận. Mặc dù đau, mềm và thường xuất hiện khối, một số di căn có thể yên lặng hình 37.

Hình 38. CT + C cho thấy một khối ngấm thuốc ở các cơ cạnh sống phải. Hình ảnh này không đặc trưng. Bệnh nhân bị di căn tới cơ từ ung thư biểu mô phổi.
(Hết. Lược dịch từ head and neck Imaging của P. Som và H. Curtin)
——————————–
Tham Khảo
61. Barnes L, Verbin R, Gnepp D. Diseases of the nose, paranasal sinuses and nasopharynx. In: Barnes L, ed. Surgical Pathology of the Head and Neck. Vol 1. New York: Marcel Dekker, 1985;725–880.
75. Patel SC, Silbergleit R, Talati SJ. Sarcomas of the head and neck. Top Magn Reson Imaging 1999;10:362–375.
89. Ginsberg L, Eicher S. Levator claviculae muscle presenting as a neck mass: CT imaging. J Comput Assist Tomogr 1999;23:538–539.
90. Rubenstein D, Escott E, Hendrick L. The prevalence and CT appearance of the levator claviculae muscle: a normal variant not to be mistaken for an abnormality. Am J Neuroradiol 1999;20:583–586.
91. Bredella MA, Tirman PF, Fritz RC, et al. Denervation syndromes of the shoulder girdle: MR imaging with electrophysiologic correlation. Skeletal Radiol 1999;28:567–572.
92. King A, Ahuja A, Leung S, et al. MR features of the denervated tongue in radiation induced neuropathy. Br J Radiol 1999;72:349–353.
93. Porter SB, Blount BW. Pseudotumor of infancy and congenital muscular torticollis. Am Fam Physician 1995;52:1731–1736.
94. Tavill MA, Wetmore RF. A case of familial sternocleidomastoid tumor of infancy. Int J Pediatr Otorhinolaryngol 1996;38:163–168.
95. Bedi D, John S, Swischuk L. Fibromatosis colli of infancy: variability of sonographic appearance. J Clin Ultrasound 1998;26:345–348.
96. Ablin D, Jain K, Howell L, West D. Ultrasound and MR imaging of fibromatosis colli (sternomastoid tumor of infancy). Pediatr Radiol 1998;28:230–233.
97. Burstein FD, Cohen SR. Endoscopic surgical treatment for congenital muscular torticollis. Plast Reconstr Surg 1998;101:20–24; discussion 25–26.
98. Haasbeek JF, Lessard JA. Isolated atlantoaxial rotatory fixation in a child with seronegative spondyloarthropathy presenting with torticollis. J Rheumatol 1998;25:169–172.
99. Svetel M, Sternic N, Filipovic S, Kostic V. Spasmodic torticollis associated with multiple sclerosis: report of two cases. Mov Disord 1997;12:1092–1094.
100. Krauss JK, Seeger W, Jankovic J. Cervical dystonia associated with tumors of the posterior fossa. Mov Disord 1997;12:443–447.
101. Entel RJ, Carolan FJ. Congenital muscular torticollis: magnetic resonance imaging and ultrasound diagnosis. J Neuroimaging 1997;7: 128–130.
102. Ackerman J, Chau V, Gilbert-Barness E. Pathological case of the month. Congenital muscular torticollis. Arch Pediatr Adolesc Med 1996;150:1101–1102.
103. Gupta AK, Roy DR, Conlan ES, Crawford AH. Torticollis secondary to posterior fossa tumors. J Pediatr Orthop 1996;16:505–507.
104. Brans J, Aramideh M, Bosch A, Speelman H. Late presentation of congenital muscular torticollis: a non-dystonic cause of torticollis. J Neurol 1996;243:354–356.
105. Maheshwaran S, Sgouros S, Jeyapalan K, et al. Imaging of childhood torticollis due to atlanto-axial rotatory fixation. Childs Nerv Syst 1995;11:667–671.
106. Milanov I, Georgiev D. Spasmodic torticollis and tremor due to multiple sclerosis: a case report. Funct Neurol 1995;10:281–285.
107. Hayashi S, Ito K, Kogure T, et al. Clinical evaluation of congenital muscular torticollis by using MR imaging. Nippon Igaku Hoshasen Gakkai Zasshi 1995;55:957–960.
108. Cammarota A, Gershanik OS, Garcia S, Lera G. Cervical dystonia due to spinal cord ependymoma: involvement of cervical cord segments in the pathogenesis of dystonia. Mov Disord 1995;10:500–503.
109. Kikuchi K, Kowada M, Kojima H. Hypoplasia of the internal carotid artery associated with spasmodic torticollis: the possible role of altered vertebrobasilar haemodynamics. Neuroradiology 1995;37: 362–364.
110. Youkilis RA, Koch B, Myer CM 3rd. Ultrasonographic imaging of sternocleidomastoid tumor of infancy. Ann Otol Rhinol Laryngol 1995;104:323–325.
111. Eysel P, Rompe JD. Osteoid osteoma of the axis. Eur Spine J 1994;3:231–232.
112. Bussieres A, Cassidy JD, Dzus A. Spinal cord astrocytoma presenting as torticollis and scoliosis. J Manipulative Physiol Ther 1994;17:113–118.
113. Singer C, Green BA, Bruce JH, et al. Late presentation of congenital muscular torticollis: use of MR imaging and CT scan in diagnosis. Mov Disord 1994;9:100–103.
114. Hladky JP, Lejeune JP, Singer B, et al. Osteoblastoma of the odontoid process. Pediatr Neurosurg 1994;21:260–262.
115. Cataltepe SU, Barron TF. Benign paroxysmal torticollis presenting as ‘‘seizures’’ in infancy. Clin Pediatr (Phila) 1993;32:564–565.
116. Yasutomo K, Hashimoto T, Miyazaki M, Kuroda Y. Recurrent torticollis as a presentation of moyamoya disease [letter]. J Child Neurol 1993;8:187–188.
117. Davids JR, Wenger DR, Mubarak SJ. Congenital muscular torticollis: sequela of intrauterine or perinatal compartment syndrome. J Pediatr Orthop 1993;13:141–147.
118. Hanko J, Hindfelt B, Matilainen T, Sjoberg S. CT-scanning and magnetic resonance imaging in idiopathic spasmodic torticollis. Acta Neurol Scand 1992;86:267–270.
119. Yasutomo K, Hashimoto T, Miyazaki M, et al. [Moyamoya disease manifested by recurrent torticollis]. No To Hattatsu 1992;24: 391–393.
120. Krauss JK, Mohadjer M, Braus DF, et al. Dystonia following head trauma: a report of nine patients and review of the literature. Mov Disord 1992;7:263–272.
121. Bredenkamp JK, Maceri DR. Inflammatory torticollis in children. Arch Otolaryngol Head Neck Surg 1990;116:310–313.
122. Isaac K, Cohen JA. Post-traumatic torticollis. Neurology 1989;39: 1642–1643.
123. Whyte AM, Lufkin RB, Bredenkamp J, Hoover L. Sternocleidomastoid fibrosis in congenital muscular torticollis: MR appearance. J Comput Assist Tomogr 1989;13:163–164.
124. Plant GT, Kermode AG, du Boulay EP, McDonald WI. Spasmodic torticollis due to a midbrain lesion in a case of multiple sclerosis. Mov Disord 1989;4:359–362.
125. Besson JA, Ebmeier KP, Gemmell HG, et al. Brain imaging and treatment response in spasmodic torticollis. Br J Psychiatry 1988;153:399–402.
126. Nicholson P, Higgins T, Forgarty E, et al. Three-dimensional spiral CT scanning in children with acute torticollis. Int Orthop 1999;23:47–50.
127. Schwarz N. The fate of missed atlanto-axial rotatory subluxation. Arch Orthop Trauma Surg 1998;117:288–289.
128. Snyder CH. Paroxysmal torticollis in infancy. A possible form of labyrinthitis. Am J Dis Child 1969;117:458–460.
129. Dunn DW, Snyder CH. Benign paroxysmal vertigo of childhood. Am J Dis Child 1976;130:1099–1100.
130. Crawford A, Harnsberger H, Johnson L, et al. Fibromatosis colli of infancy. AJR 1988;151:1183–1184.
131. Maddalozzo J, Goldenberg JD. Pseudotumor of infancy—the role of ultrasonography. Ear Nose Throat J 1996;75:248–254.
132. Mann SS, Som PM, Gumprecht JP. The difficulties of diagnosing myositis ossificans circumscripta in the paraspinal muscles of a human immunodeficiency virus–positive man: magnetic resonance imaging and temporal computed tomographic findings. Arch Otolaryngol Head Neck Surg 2000;126:785–788.
133. Kransdorf MJ, Meis JM, Jelinek JS. Myositis ossificans: MR appearance with radiologic-pathologic correlation. AJR 1991;157: 1243–1248.
134. Steiner M, Gould AR, Kushner GM, et al. Myositis ossificans traumatica of the masseter muscle: review of the literature and report of two additional cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997;84:703–707.
135. Brooke M. Disorders of skeletal muscle. In: Bradley W, Daroff R, Fenichel G, Marsden C, eds. Neurology in Clinical Practice. Vol 2. Boston: Butterworth-Heinemann, 1991;1843–1886.
136. Wilson A, Mackay L, Ord R. Recurrent dislocation of the mandible in a patient with myotonic dystrophy. J Oral Maxillofac Surg 1989;47:1329–1332.
137. Vanier T. Dystrophia myotonica in chilhood. Br Med J 1960;2:1284–1288.
138. Kawamura T. Computed tomographic findings of brain and skull in myotonic dystrophy, [letter]. J Oral Maxillofac Surg 1987;50: 1723.
139. Cornacchia L, Marina R, Ballanini V, Sozzi G. Computed tomographic findings of brain and skull in myotonic dystrophy [letter]. J Oral Maxillofac Surg 1988;51:1463–1464.
140. Saitoh H. [CT findings of muscular dystrophy: limb girdle type (LG), myotonic type (MYD) and Duchenne type (DMD)]. Nippon Igaku Hoshasen Gakkai Zasshi 1991;51:790–798.
141. Som P, Rothschild M, Silvers A, Norton K. A painless retroauricular mass in a patient with myotonic dystrophy: computed tomographic documentation of the bone changes that occur in the skull base. J
Skull Base Surg 1998;7:223–225.
142. Lee J, Lee M, Lee B, et al. Rhabdomyosarcoma of the head and neck in adults: MR and CT findings. Am J Neuroradiol 1996;17:1923–1928.
143. Resnick D, Niwayama G. Skeletal metastases. In: Resnick D, ed. Diagnosis of Bone and Joint Disorders. Vol 6. 3rd ed. Philadelphia: WB Saunders, 1995;4039–4064.